

Interactive Mutation Browser
Interactive Mutation Browser
Prototyping a new way to interact with Protein Language Models

Welcome to The Century of Biology! This newsletter explores data, companies, and ideas from the frontier of biology. You can subscribe for free to have the next post delivered to your inbox:
This post is an experiment in sharing a new research result, so it’s a bit more on the technical side of the spectrum of posts you’ll be receiving from me.
Enjoy! 🧬

In many ways, AI has already dramatically changed my life. The emergence of powerful Foundation Models for natural language has sparked a software renaissance. I’ve infused countless new tools into my research, writing, and coding workflows. There’s simply never been a better time to be trying to learn about things. For research, I use Explainpaper to dive into new studies that I have less context for, and Elicit to supercharge literature search. Tools like Github Copilot and ChatGPT have irreversibly transformed my approach to programming.
All of this innovation has made me wonder: where are the new tools for biologists?
As the saying goes, be the change you wish to see in the world. Here, I’m going to share an attempt I’ve made to design a new type of interaction with Protein Language Models: the Interactive Mutation Browser. This essay is a new experiment in sharing this small research result here on Substack.
Why am I deciding to share it here?
The main currency of academic science is the publication. As I’ve previously written about, there are some real challenges in fitting in to this system for researchers whose primary output is software. To be somewhat blunt, scientific software is often treated as a second-tier output, and focusing on making it is a pretty bad academic career decision.
It’s worth being specific about how this works. If you build a new software tool, you need to write a short paper called an Application Note to score a few academic points for it. These are peanuts compared to a full Journal Article, but still something. The last time I did this, the paper was rejected by the bioRxiv preprint server—which is frankly impressive given their intended ~100% acceptance rate—because a useful new software package is not considered a research result in its own right. Finally, the paper saw the light of day, and now a lot of people use and enjoy the tool.
So, in an attempt to circumvent this process, here’s the director’s cut explaining why I decided to build this new tool, and what it does.
UI for AI
*Record Scratch* *Freeze Frame* So, how did I end up writing scientific software in the first place?
One way to classify a research project is to stratify the type of risk involved in the project. A useful example of this is to estimate the amount of scientific risk vs. the amount of engineering risk you’re committing to in order to reach success.
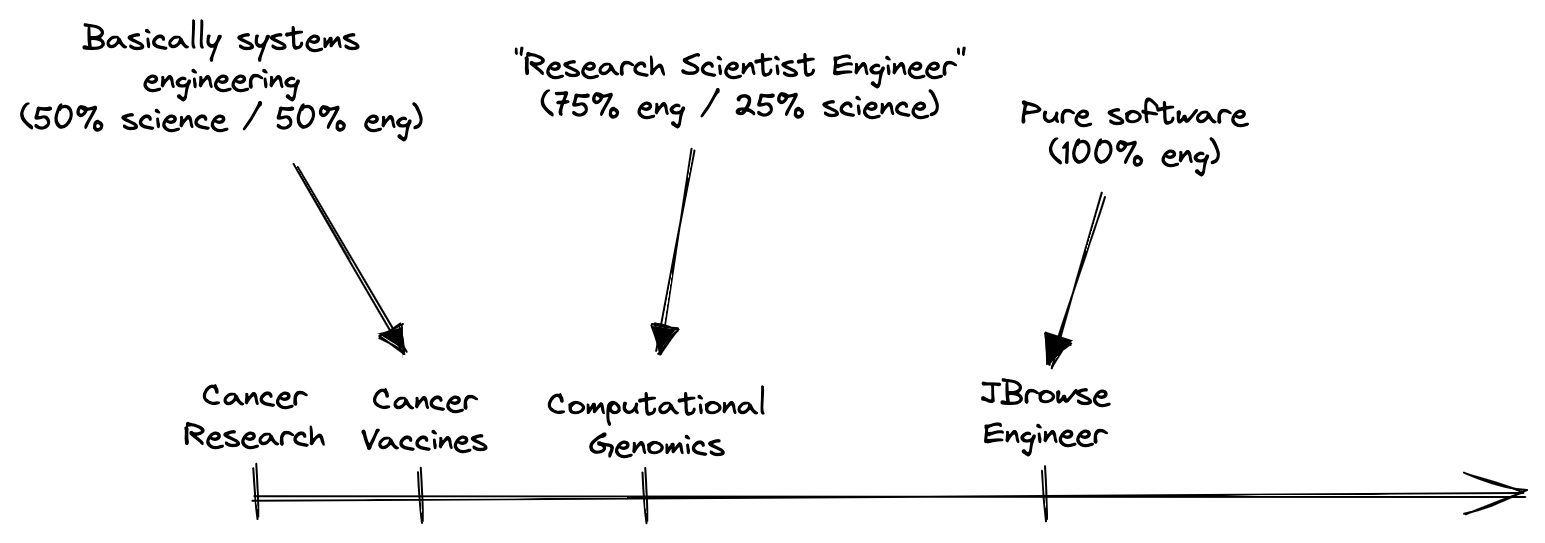
I started out my scientific career working on projects with 100% scientific risk and 0% engineering risk. My first research projects used extremely well-established molecular biology tools to answer new questions about the underlying genetics of cancer metastasis. There was nothing novel about the approach, the major risk was whether or not we would find interesting results.
, and Sid Pradeep, Class of 2017 (right).")
Over time, the pendulum started to swing farther towards engineering. I was more intellectually interested in solving systems-level research bottlenecks by building new tools than any single basic science question. I transitioned from cancer research to cancer vaccine development, and then to a “Research Scientist Engineer” job working on computational genomics. I was about to start graduate school and swing back in the science direction when I got nerd sniped by this tweet from Ian Holmes1:

I had previously built a scientific Web app for a project, and felt that there was a lot of daylight between the design ideas I had for the user interface, and what I was able to implement with the framework I was using. The JBrowse job was a chance to learn the tools and techniques that gave people the power to build whatever they want on the Web, while still directly working on genomics. Sign me up! So, I spent a year becoming a React engineer and learning how to make advanced scientific user interfaces.
At this point, I headed off to graduate school and spent time thinking about what to work on. I had the weird skillset of a trained biologist who also happened to be a React engineer. Were there any unique openings for me to explore? Among several research directions that I’ve embarked upon, one observation tugged at me: there seemed to be a sizable gap between the BioML and BioViz communities:
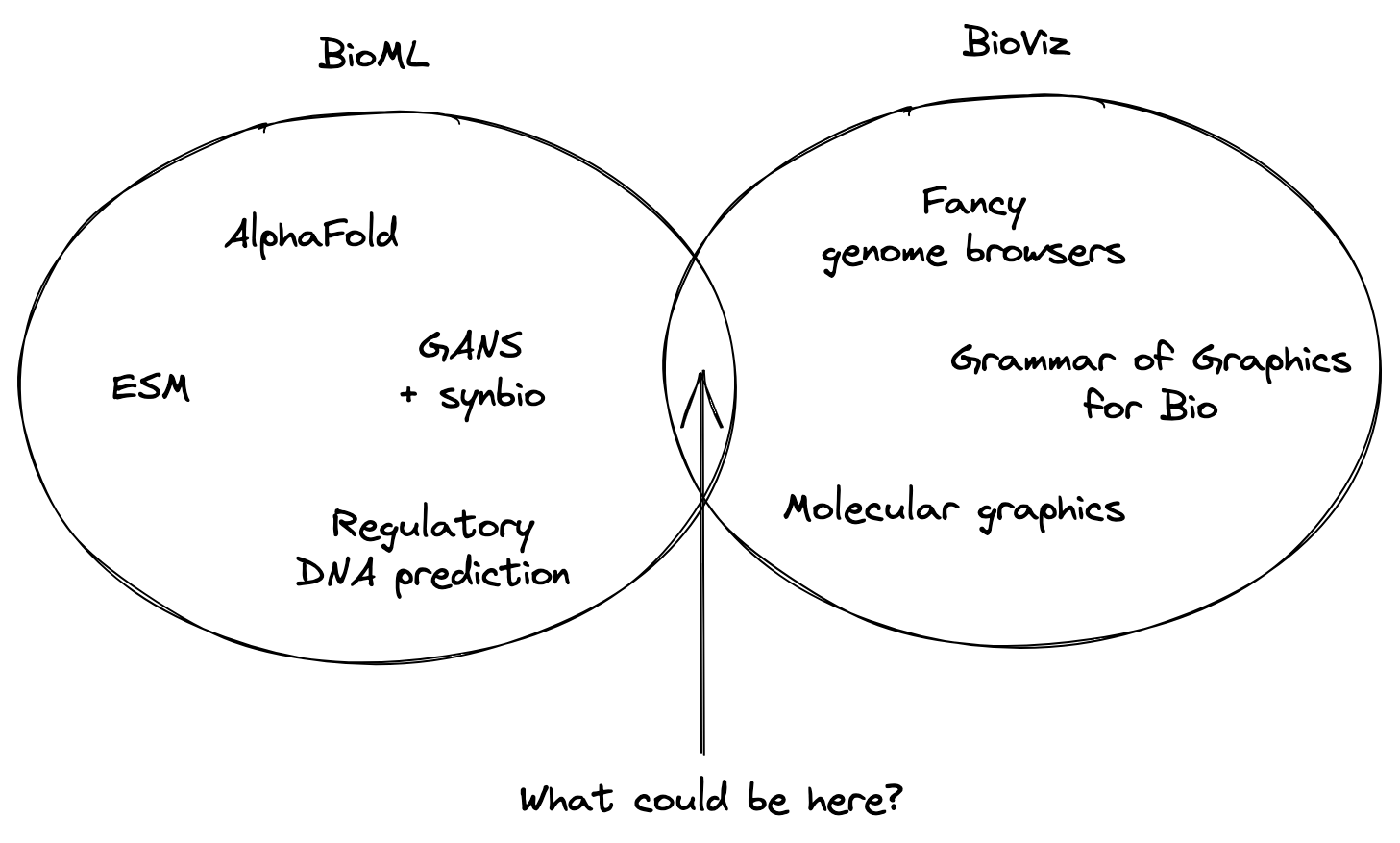
I’d been excited about ML for biology since I worked on epitope prediction for cancer vaccines during undergrad, but over the last decade things have really started to take off for this field. It’s been exciting to see some of the most substantial ML advances happen in the life sciences. At the same time, a small but dedicated community of researchers has continued to research and build new visualization technologies for biologists—like the types of genome browser tools we built at JBrowse, to a new grammar for genomic visualization, and the tools for interactively viewing molecules in a Web browser.
However, these fields haven’t collided in a real way yet.
ML researchers are primarily content with their research output coming in the form of papers and Github repositories with model weights.2 Visualization researchers have been happy to continue building tools for interacting with static data generated from research instruments. But what might be possible when we start to build new UI for AI?
This isn’t a trivial academic niche—one of the major leaps forward for OpenAI has been the development of the chat interface that provides a new type of experience for interacting with models. As the explosion of tools like Explainpaper, Elicit, Perplexity, and Bearly are showing us, we’re only scratching the surface of the design space for model interaction.
In the parlance of the very wise design researcher Don Norman, these new models significantly change the technical constraints that app builders are working with. It’s now possible to rapidly generate succinct explanations of complex text using Large Language Models (LLMs), or protein structure predictions using Protein Language Models (PLMs). The advent of modern Web search changed our relationship to information. Now, we have these strange and beautiful compressed representations of language—both human language and protein language. How can we learn to interact with these new oracles and uncover the new knowledge they are capable of guiding us to?
When the constraints change, it becomes possible to design new affordances, which are “a relationship between the properties of an object and the capabilities of an agent that determine just how the object could possibly be used.” Put simply, we need to imagine new interfaces for these powerful models to make use of all of the newfound capabilities that they provide us. What types of apps are now possible to build with these new types of practically instantaneous generative and predictive capabilities? It’s a genuine research problem. I’ve been inspired by researchers like Linus Lee, who have been tackling this problem head-on. Here’s how he describes his work:
My research investigates the future of knowledge representation and creative work aided by machine understanding of language. I prototype software interfaces that help us become clearer thinkers and more prolific dreamers.
He’s publicly experimented with all new types of UI interactions, like creating a latent space for your thoughts by making it possible to shrink and expand sentences by simply dragging the text with your mouse.3 Again, all of this cool experimentation has made me wonder…
What new interfaces and experiences are possible for biologists?
FM-Powered Apps
As I stewed on all of this, I had to finish the rest of my graduate school courses. For my computational requirement, I decided to take a course called Advances in Foundation Models.4 Given that it was taught by some of the professors that coined this term, it seemed like a pretty great opportunity to understand some of the ideas behind the insane models like GPT and to learn about what might be coming next. At this point, you might be asking yourself: what is a Foundation Model (FM)?
The core idea is that a FM is a deeply general AI model, trained on massive data, that can be used for a large and diverse set of downstream tasks:
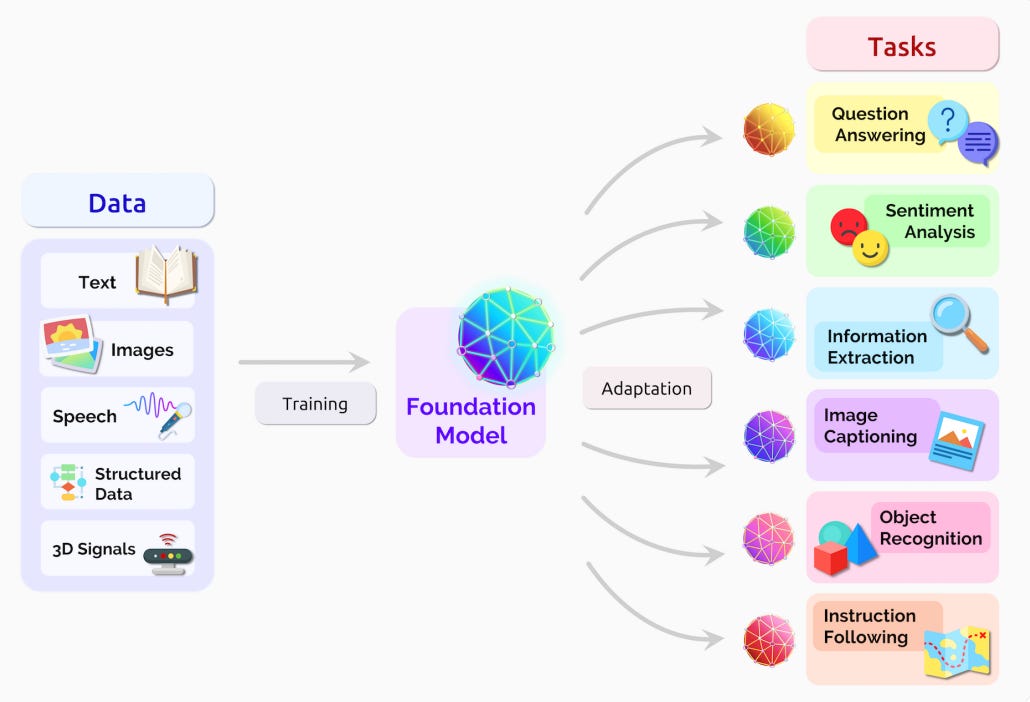
There is a general trend in AI towards homogenization—instead of training bespoke models for each specific task, it’s often better to tune extremely powerful (and large) FMs with general language or image capabilities with a little bit of additional data or prompting.
On a technical level, there are two major driving factors that have made FM models possible: transfer learning, and scale.
Transfer learning is an important subfield of ML focused on making it possible to repurpose previously trained models to new tasks by reapplying the knowledge that has already been learned through large-scale training. It lets you train a giant image model, and then fine tune it to recognize a specific set of objects extremely well using a smaller additional data set.5
Modern hardware and new model architectures have made it possible to train models at an entirely new scale, utilizing Internet-scale data. A fairly non-obvious result of this new model scale is that they have developed emergent capabilities. A good example is the emergence of the ability to apply a model to new downstream tasks with only natural language prompting and no re-training. It can be easy to lose track of the pace of AI progress, but this only became possible in the transition from GPT-2 to GPT-3.6
As the authors of the FM paper say, “Transfer learning is what makes foundation models possible, but scale is what makes them powerful.”
Alright, now we have more context for what FMs are. In this course, I had the option to create a “FM-powered application” of my choosing. This is an application built to interact with model predictions in real time to accomplish a new task. This made me ask the most immediate question: what is the clearest example of a bioFM right now?
To me, the most obvious success in BioML so far has happened at the interface of protein sequence and structure. I’ve written about this several times before, but I view the AlphaFold moment—effectively solving the sequence-to-structure prediction problem—as a real breakthrough for AI in the life sciences.
So, what type of application is now possible because of Protein FMs?
The Interactive Mutation Browser
One of the most powerful new protein models is ESM, created by Meta AI. ESM is a Protein Language Model that can be used for a variety of downstream tasks including extremely fast structure prediction from a single amino acid input sequence. Meta recently used ESM to create a new atlas of more than 600 million predicted protein structures from metagenomic sequencing data.
Recently, a research group at UCSF used ESM to do genome-wide variant effect prediction—providing further evidence that it truly has FM-like properties. I’ve previously written about this work in detail, which hopefully serves as a good starting point for understanding PLMs.

Variant effect prediction is an important problem in computational genetics. The holy grail is to have a computational system that can take any genetic variant—a DNA mutation—and predict what the impact will be on downstream biological function. This recent study used ESM to predict the impact of every single possible missense mutation in the genome, which is a mutation that results in an amino acid change in a protein.
Essentially, if you change an amino acid in a protein sequence, can you guess what will happen to the protein structure? Will it change the structure in a way that disrupts its function?
Here’s an example of the type of molecular detective work that the authors did in this study:
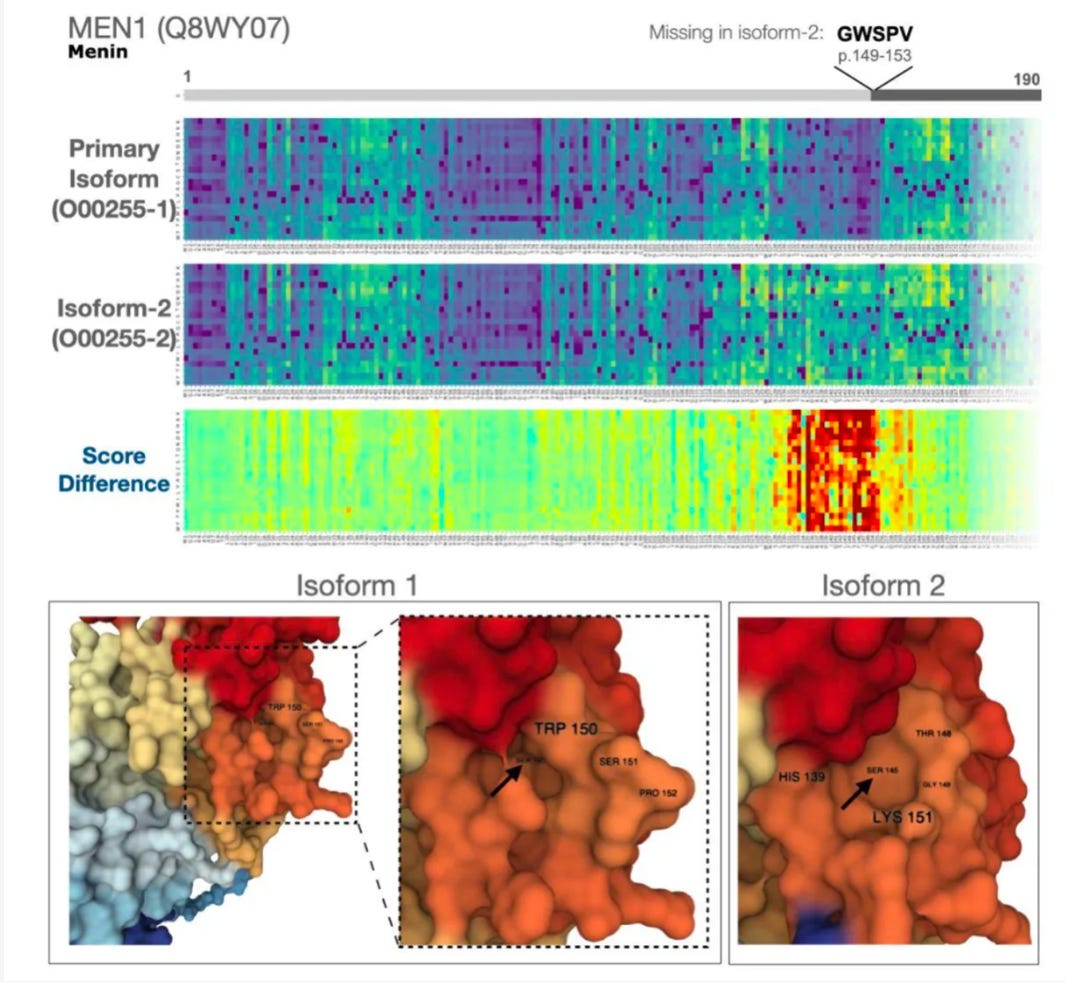
You can see the isoform is missing the amino acids GWSPV starting at residue 149. This messes up a small binding pocket on the protein surface. Variants in this region had been previously reported to be associated with cancer, but it was hard to know what was actually happening mechanistically.
I viewed this as a clear starting point for a new tool. This study tested every individual missense variant, but predicting the impact of every single possible combination of mutations or potential insertion or deletion is combinatorially impossible. What if there could be a way to quickly specify any possible change to an amino acid and immediately see the predicted structural change?
The core idea of the Interactive Mutation Browser is that Protein FMs now make this possible.
The workflow starts by entering an amino acid sequence. Next, I created a simple interactive mutation editor to make any set of changes to the sequence, including amino acid substitutions, insertions, or deletions:
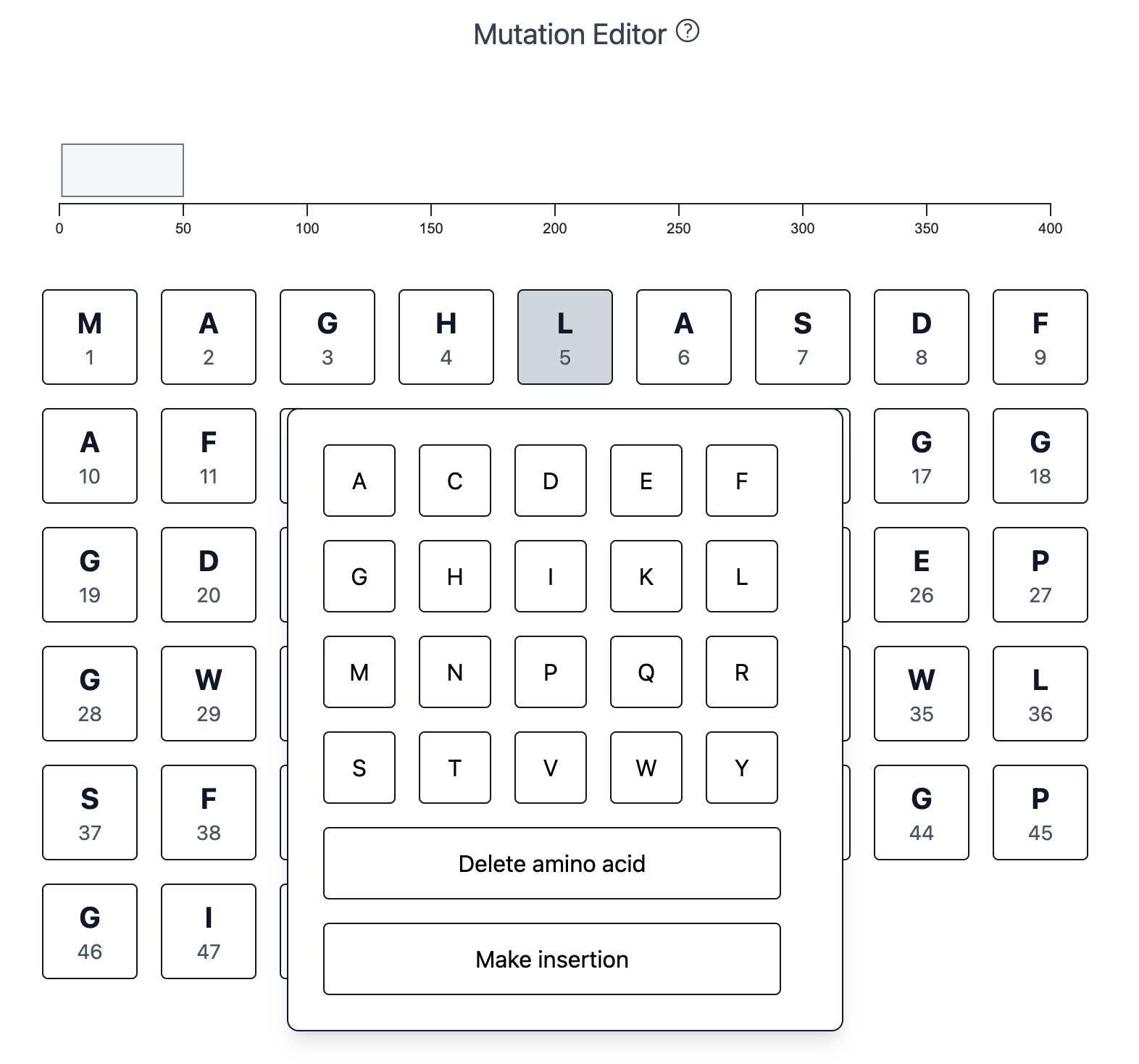
When you make a change, the sequence-wide browser track records it for you:
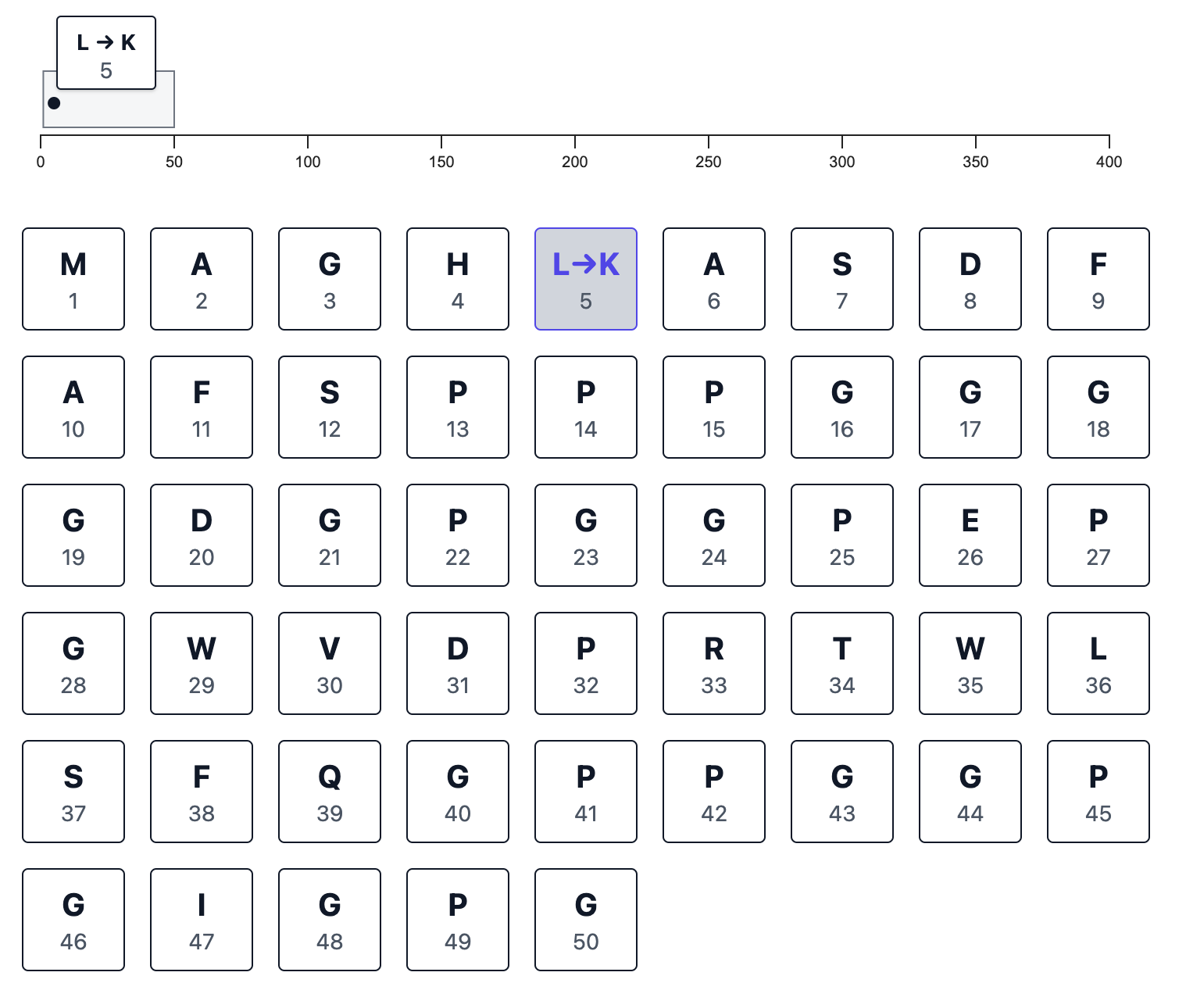
Next, you click a single button and immediately get an interactive visualization of the wild-type and mutated protein structures side-by-side. The changed residues are highlighted in red—and a stick model is also used to help draw attention to any changes. Below, I’m showing an example for a very short sequence of amino acids, where the changes are immediately visually detectable without any zooming or panning.

What’s actually happening under the hood? The sequences from the editor are being sent to the ESM Atlas API, which sends back the structure predictions—typically in a matter of seconds. Next, these structures are loaded into 3Dmol.js molecular viewers—which are built using WebGL technology, enabling fast and performant 3D graphics in a Web browser.
In other words, here’s the answer to my previous question:

My hope is that this tool helps enable molecular exploration and model interpretability. By being able to quickly test the relationships between inputs and outputs, we can gain intuition for what the models are good at, as well as the different ways that they don’t work as well. As the models continue to improve, it should serve as a new type of workbench for in silico protein surgery.
Limitations, Related Work, Future Directions
As a research prototype, there are many ways that this tool could be made better. To start, I used the ESM Atlas API for this project, because I mainly wanted to focus on design research instead of hassling with deploying and hosting my own ML model. The main drawback with this choice is that the API doesn’t accept input sequences longer than 400 amino acids. The UCSF group solved this length restriction problem in their work, but there isn’t a publicly available API for that model.
The mutation editor is also intentionally as simple as possible. I decided to focus on amino acid sequences, but a DNA editor would also be a nice feature. Accepting variant file types like VCF would also add flexibility. Clearly, a lot of room for improvement on this front!
I’m also not convinced that the side-by-side view is the best possible visualization experience. An alternative would be to create a superposition of the two structures, which could quickly draw visual attention to regions where they differ. It could also be valuable to include a statistic or visualization of the confidence prediction for the predicted structure.
So, if you’re a hacker who wants to help me out with this project, here are some tangible ideas:
ML improvements — help out by hosting the length unrestricted ESM model, ideally returning both structure predictions and confidence predictions.
Front-end improvements — help out by exploring ways to make the mutation editor a more full-featured and flexible system.
Molecular graphics improvements — help out by experimenting with new ways to visualize the wild-type and mutated structures together. One option could be to use a different library, like Mol*, which supports superposition views and may offer a more full-featured experience.
The code is open source, and I’d be more than happy to explore co-authorship with anyone interested in collaborating on this if it turns into a short publication! Please reach out if interested.
Part of being a good researcher is understanding the current space of work and ideas related to your project. There was a cool preprint introducing a tool with similar ideas called MutationExplorer. At first I saw the title and thought… dang. When I took a closer look I got really excited. This group used the Rosetta Software Suite for structure predictions. Now, you can compare the results of both modeling approaches right next to each other in the browser. How cool is that??
I’m also not the first person to experiment with linking structure predictions to interactive graphics viewers. The software package gget has a feature that lets you immediately see the AlphaFold structure prediction inside of a Jupyter notebook by running a single Python function. Clearly this idea is in the air right now, because Benchling also recently added an AlphaFold viewer to their product using Mol*.
As an academic, I view it as my role within the ecosystem to experiment with novel types of interactions and affordances that expand our working relationship with these models. I take some inspiration here from Andy Matuschak, who has spent time as an independent researcher after helping to build iOS at Apple. Some of his new UI ideas have trickled into several software products. Similarly, if this idea takes off, I would love to someday see a mutation editor inside of a product like Benchling, Latch, or somebody else!
Finally, I think that this tool is just scratching the surface of what is possible at the interface of BioML and BioViz. For an early Christmas present (published December 22, 2022) last year, the Meta AI team dropped a preprint describing the specification for a high-level programming language for generative protein design. This is the type of thing that makes me pinch myself because of how lucky I feel to be alive right now working on computational biology.
What kind of interface could be designed for protein designers using this type of language?
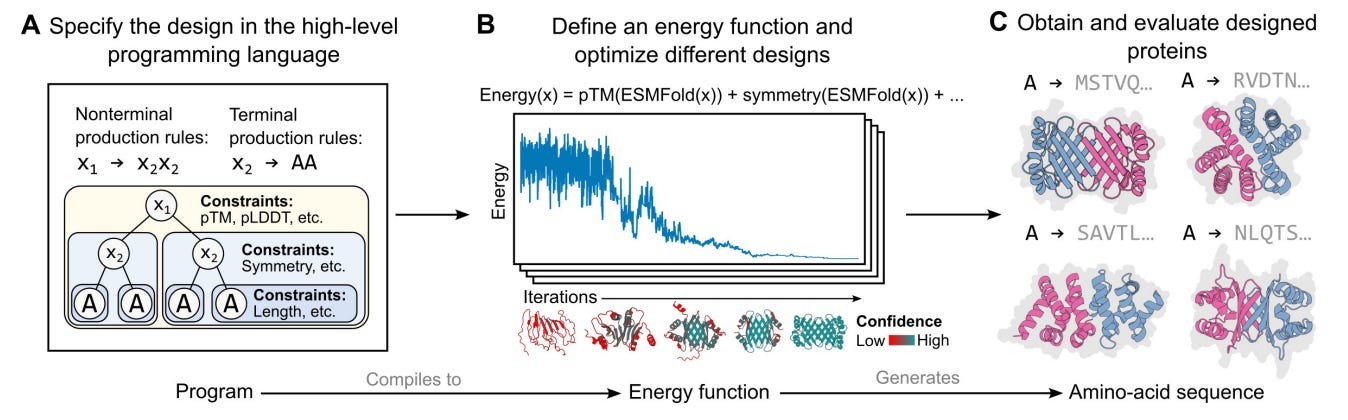
Immediately, it seems like there are connections that could be made to interfaces for visual programming languages. Imagine interactively building out the graph of protein constraints on one half of your screen, while a 3D protein viewer continually updates on the other half. This is just one possible idea for bringing this language out into the world for scientists to use and benefit from.
Hopefully this new tool and essay helps to get you thinking about the following question:
How can we use AI to give biologists superpowers?
Thanks for reading about the Interactive Mutation Browser — a new experiment in interacting with Protein Language Models.
I want to thank David Koes at the University of Pittsburgh for building and maintaining 3Dmol.js, and for fielding my questions as I experimented with it. I’d also like to thank the Meta team for releasing a public API for the ESM model, making this project much easier to embark on.
Thanks to my advisor, Michael Fischbach, who gave me the freedom to explore this side project and supported me in this experiment to share it with you here.
Also, thanks to
for reading a draft to help me gage how well I explain this tool to a smart and curious general audience. If something doesn’t make sense, blame him, not me!Thank you to Kelda for proof-reading and editing.
To be fair, I was deciding between this awesome job and an entirely virtual first year of graduate school during the height of the COVID pandemic. It was an easy choice!
In the best case scenario. Still, too many ML papers don’t share models or code.
If scientific software is second-tier output in academia, this type of experimentation would be viewed as third-tier. Most academics—especially academic biologists—probably wouldn’t call this research. Despite that, I think that this type of work will be important to reach the full promise of computational biology, and we can’t expect this to magically happen somewhere outside of the ivory tower.
Many of the guest lectures are freely available through the awesome Stanford MLSys Seminars YouTube channel. Check it out!
On a more technical level, this involves freezing the weights of the earlier layers in the network, that have learned more general properties of shapes within images, and then re-training the deeper layers closer to the output with new and more specific data. One of the great AI educators, Jeremy Howard of fast.ai, has been teaching people how to do amazing things with transfer learning for years.
This was a particularly big scale-up, going from 1.5 billion parameters to 175 billion parameters.
Really nice stuff, Elliot. I think this is your best piece yet. Love the fusion of research results with general-facing writing.
This is great! Did you know you can download Microsoft Edge browser and basically get ExplainPaper for free? (Just open a PDF in Edge and it can explain sections for you, just like ExplainPaper)